В патогенезе кишечных расстройств при муковисцидозе играет роль

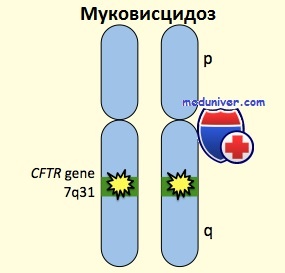
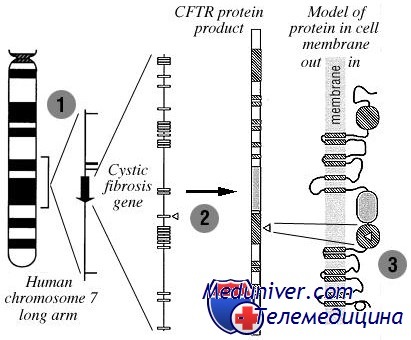
Муковисцидоз: причины, диагностика, лечениеЭтиология и встречаемость муковисцидоза. Муковисцидоз (MIM №219700) — аутосомно-рецессивное заболевание эпителиального ионного транспорта, вызываемое мутациями в гене трансмембранного регулятора муковисцидоза (CFTR). Хотя муковисцидоз бывает во всех расах, преимущественно это болезнь северных европейцев. Встречаемость муковисцидоза среди живорожденных меняется от 1 на 313 среди жителей штата Южная Альберта в Канаде до 1 на 90 000 в азиатской популяции Гавайских островов. В США у европеоидов встречаемость составляет 1 на 3200. Патогенез муковисцидозаБелок CFTR формирует регулируемый циклическим аденозинмонофосфатом (цАМФ) канал транспорта хлоридов, от которого зависят другие ионные каналы. CFTR поддерживает гидратацию секрета в дыхательных путях и протоках желез, обеспечивая транспорт хлоридов и ограничивая потерю натрия. Дисфункция CFTR может влиять на множество разных органов, особенно выделяющих слизистый секрет, включая верхние и нижние дыхательные пути, поджелудочную железу, желчевыводящую систему, мужские половые органы, кишечник и потовые железы. Густой и вязкий секрет в легких пациентов с муковисцидозом создает помехи в мукоцилиарном пространстве, тормозит функцию естественных антимикробных пептидов, обеспечивает среду для роста патогенных микроорганизмов и затрудняет прохождение воздуха. В течение первых месяцев жизни в этот секрет заселяются бактерии, вызывающие воспалительную реакцию. Выброс провоспалительных цитокинов, антибактериальных ферментов, а также бактериальных ферментов повреждает бронхиолы. Повторные циклы инфекции, воспаления и уничтожения ткани уменьшают количество функциональной ткани легкого и, в конечном счете, приводят к дыхательной недостаточности. Нарушение транспорта хлоридов в поджелудочном протоке усиливает вязкость секрета и приводит к задержке пищеварительных ферментов в поджелудочной железе, что, в конечном счете, вызывает фиброз поджелудочной железы. CFTR также регулирует транспорт натрия и хлоридов в поте. При отсутствии функционирующего CFTR пот имеет высокое содержание хлоридов и натрия. Это лежит в основе исторического названия — «синдром соленого ребенка» и диагностического теста на хлориды пота.
Фенотип и развитие муковисцидозаКлассически муковисцидоз обнаруживают в раннем детстве, хотя приблизительно 4% случаев диагностируют во взрослом возрасте; 15-20% больных имеют при рождении мекониальный илеус, у остальных позже появляются хронические респираторные жалобы (риниты, синуситы, обструктивная болезнь легкого) или задержка роста, или и то, и другое. Задержка роста вызвана сочетанием повышенного расхода энергии из-за хронических легочных инфекций и нарушением всасывания из экзокринной недостаточности поджелудочной железы. От 5 до 15% пациентов с муковисцидозом не имеют поджелудочной недостаточности. У более 95% мужчин с муковисцидозом отмечают азооспермию из-за врожденного двустороннего отсутствия семявыносящего протока. Развитие легочной патологии — главная причина смертности и инвалидности. Большинство пациентов умирают от дыхательной и правожелудочковой недостаточности, вызванной гибелью паренхимы легких и высоким легочным сопротивлением (cor pulmonale); средний возраст выживания в Северной Америке — 33 года. Помимо муковисцидоза, мутации в гене CFTR связаны с целым спектром болезней, включая обструктивную азооспермию, идиопатические панкреатиты, распространенные бронхоэктазы, аллергический бронхолегочный аспергиллез и бронхиальную астму. Некоторые из этих болезней вызваны мутациями в одном из аллелей CFTR; другие, подобно муковисцидозу, наблюдают, только если мутации присутствуют в обоих аллелях CFTR. Прямая причинно-обусловливающая роль мутантных аллелей CFTR доказана для некоторых, но не всех перечисленных заболеваний. Корреляция между конкретными мутантными аллелями CFTR и тяжестью болезни существует только для недостаточности поджелудочной железы. Второстепенные мутации или полиморфизмы в пределах аллеля CFTR могут изменять эффективность сплайсинга или созревания белка, тем самым расширяя спектр патологии, связываемой с некоторыми мутациями. Кроме того, некоторые мутации в CFTR вызывают проявления болезни только в определенных тканях; например, некоторые мутации, нарушающие эффективность сплайсинга, имеют больший эффект в производных вольфовых протоков, чем в других тканях, вследствие тканеспецифичной потребности в транскрипте и белке полного размера. Факторы влияния окружающей среды, например воздействие табачного дыма, заметно усиливают тяжесть патологии легких у пациентов с муковисцидозом. Особенности фенотипических проявлений муковисцидоза:
Лечение муковисцидозаПоскольку в гене CFTR описано более 1000 различных мутаций и вариантов, диагноз «муковисцидоз» в США обычно основывается на клинических критериях и концентрации хлоридов пота. У 1-2% пациентов с муковисцидозом концентрация хлоридов пота оказывается в пределах нормы; тем не менее обычно муковисцидоз у этих пациентов позволяют диагностировать аномальные результаты измерений различия трансэпителиальных потенциалов слизистой оболочки носа. В настоящее время полное излечение муковисцидоза невозможно, хотя симптоматическое лечение повысило среднюю продолжительность жизни с раннего детства до 30-40 лет. Цели медицинской помощи при муковисцидозе — выведение легочного секрета, лечение легочных инфекций, заместительная терапия ферментами поджелудочной железы, достаточное питание и предотвращение кишечной непроходимости. Хотя лечение замедляет развитие легочной патологии, единственное эффективное лечение дыхательной недостаточности при муковисцидозе — пересадка легкого. Заместительная терапия ферментами поджелудочной железы и добавление жирорастворимых витаминов эффективно помогают в лечении мальабсорбции; тем не менее повышенная энергетическая потребность и анорексия у многих пациентов требуют также дополнительного поступления калорий. Большинство пациентов также нуждаются в расширенном консультировании, у многих отмечают психологические эффекты наличия хронического тяжелого заболевания. В 2004 г. Американский колледж медицинской генетики, Центры США по лечению и профилактике заболеваний и Фонд муковисцидоза рекомендовали введение неонатального скрининга на муковисцидоз, поскольку обнаружение болезни в периоде новорожденности позволяет предотвратить нарушения питания, наблюдаемые у клинически недиагностированных пациентов с недостаточностью поджелудочной железы. Отдаленное влияние на выживаемость и течение легочной патологии пока неясно. Риск наследования муковисцидозаЭмпирический риск для пары иметь ребенка, больного муковисцидозом, существенно меняется в зависимости от частоты муковисцидоза в разных этнических группах. Для жителей США — выходцев из Северной Европы без случаев муковисцидоза в семье эмпирический риск для каждого родителя оказаться носителем составляет приблизительно 1 на 29, следовательно, риск иметь больного ребенка — 1 на 3200. Для семейных пар, уже имеющих ребенка с муковисцидозом, риск для последующих детей иметь муковисцидоз — 1 к 4. В 1997 г. конференция Национального института здоровья выработала консенсус, рекомендующий предлагать тестирование на носитель-ство муковисцидоза всем беременным и семейным парам, планирующим беременность. Американский колледж акушерства и гинекологии подтвердил эти рекомендации. По данным 2004 г., до 25% беременных в США проходят тестирование на носительство муковисцидоза. Пренатальную диагностику проводят методом идентификации мутаций гена CFTR в ДНК, полученной из тканей плода, например ворсин хориона или амниоцитов. Для эффективного выявления больных плодов обычно требуется, чтобы мутации, вызывающие муковисцидоз в семье, уже были известны. Пример муковисцидоза. Ж.Б., 2,5-летний мальчик, направлен в педиатрическую клинику для оценки причин задержки роста. В раннем детстве у ребенка отмечали понос и колики в животе, что разрешилось после замены детской смеси. Когда в рацион добавили обычную пищу, у ребенка появился зловонный стул, содержащий непереваренные частицы пищи. На втором году жизни мальчик плохо рос, у него развился хронический кашель и частые респираторные инфекции. В семье ну у кого не было низкого роста, нарушений питания или легочных заболеваний. При медицинском осмотре масса тела и рост ребенка оказались ниже 3-го процентиля, а окружность головы в 10-м процентиле. Отмечены выраженный пеленочный дерматит, рассеянные хрипы и небольшое расширение ногтевых фаланг. В остальном данные осмотра нормальные. После краткого обсуждения возможных причин болезни педиатр назначил несколько тестов, включая определение концентрации хлоридов пота с ионофорезом пилокарпина; уровень хлоридов пота оказался 75 ммоль/л (в норме <40 ммоль/л; неопределенные значения — 40-60 ммоль/л), что соответствует муковисцидозу. На основе этого результата и клинических находок педиатр расценил заболевание ребенка как муковисцидоз. Пациент и его родители направлены в клинику муковисцидоза для дальнейшего консультирования, определения мутации и лечения. – Также рекомендуем “Врожденная глухота – тугоухость: причины, диагностика, лечение” Оглавление темы “Наследственные болезни”:
|
Источник
МУКОВИСЦИДОЗ (лат. mucus слизь + viscidus липкий, клейкий; син.: врожденная энтеробронхопан-креатическая диспория, кистозный фиброз поджелудочной железы, фиброкистозная болезнь поджелудочной железы, панкреатическая врожденная стеаторея) — наследственная болезнь, характеризующаяся системным поражением желез внешней секреции (экзокринных желез), проявляющаяся тяжелым расстройством функции органов дыхания, желудочно-кишечного тракта и ряда других органов и систем.
В 1936 г. Г. Фанкони и в 1938 г. Андерсен (D. Н. Andersen) обнаружили у детей, умерших от тяжелых нарушений функции жел.-киш. тракта, специфические патол, изменения в поджелудочной железе. В 1944 г. Фарбер (S. Farber) установил, что многие симптомы заболевания можно объяснить дефектом слизистого секрета экзокринных желез и предложил название болезни «муковисцидоз». По данным ди Сант-Аньезе (P. A. di Sant’Agnese, 1969), частота заболевания составляет примерно один случайна 2000 новорожденных. Введение широкого обследования новорожденных на М. с помощью скрининг-программ (см. Скрининг) дало возможность улучшить раннюю диагностику заболевания и приблизиться к установлению его истинной частоты [по данным Стефана (U. Stephan, 1974), она составляет один случай на 1000 новорожденных].
Этиология и патогенез
Основной ферментативный дефект при М. неизвестен. Имеются доказательства, что это наследственная болезнь, передаваемая по аутосомно-рецессивному типу. По данным М. И. Рейдермана (1974), от 2,6 до 3,6% населения СССР являются гетерозиготными носителями этого заболевания. У родителей и ближайших родственников больных М. отмечаются хрон, бронхо-легочные и жел.-киш. заболевания, панкреатит, рассматриваемые нек-рыми клиницистами как «малые» формы М.
В основе патогенеза М. лежит секреция экзокринными железами вязкой слизи с нарушенными физ.-хим. свойствами (возрастание концентрации электролитов и белков при уменьшении водной фазы). При задержке слизистого секрета в поджелудочной железе (см.) возникает ее атрофия с последующим фиброзом. Стадия полного рубцевания поджелудочной железы может наблюдаться у детей уже в возрасте 2—3 лет. Возникновение склероза в поджелудочной железе и других органах у больных М. обусловлено не только механическим застоем слизистого секрета и затруднением его оттока; более важное значение имеют процессы нарушения обмена в формирующейся соединительной ткани, нарушение функции фибробластов — основного функционирующего элемента незрелой соединительной ткани (накопление в клетках кислых мукополисахаридов — гликозаминогликанов, связывание метахро-матического вещества с мембранами клеток).
Патологическая анатомия

Рис. 1. Микропрепараты поджелудочной железы: а — в норме; б — при муковисцидозе (1 — кистозно расширенные протоки, содержащие глыбки уплотненного слизистого секрета; 2 — прослойки соединительной ткани); окраска гематоксилин-эозином.
При муковисцидозе поражается не только поджелудочная железа, но и слизистые железы органов дыхательной, пищеварительной системы, слюнные, потовые и слезные железы. При далеко зашедшем процессе отмечается значительная плотность поджелудочной железы, дольки ее мелкие, расстояния между ними увеличиваются, иногда могут быть мелкие кисты. Изменения могут варьировать от кистозно расширенных единичных выводных протоков и ацинусов до кистозного перерождения всей экскреторной железистой паренхимы. Микроскопически в кистозно расширенных выводных протоках, вставочных протоках и в ацинусах железы наблюдается скопление густого, эозинофильного, иногда слоистого, слизистого секрета; вокруг кистозно расширенных выводных протоков, внутри и вокруг долек отмечается разрастание соединительной ткани (рис. 1), в к-рой встречаются участки интенсивной клеточной инфильтрации, состоящие из лимфоцитов, плазматических клеток и гистиоцитов. Железистая паренхима атрофична. Панкреатические островки (инсулярный аппарат) не изменены. В кишечнике (см.) изменение характера слизистого секрета наблюдается в дуоденальных (бруннеровых) железах (gll. duodenales) и в бокаловидных клетках слизистой оболочки преимущественно тонкой кишки. Клейкий вязкий слизистый секрет вызывает уплотнение, склеивание и сгущение мекониальных и каловых масс, что наряду со сте-атореей способствует копростазу, к-рый может привести к илеусу с изъязвлением, перфорацией стенки кишки и развитием мекониального или калового перитонита (см.). При мекониальном перитоните на висцеральной и париетальной брюшине новорожденного, кроме комочков мекония, отмечаются фибринозные или фибринозно-гнойные наложения. При тяжелой форме М. плода мекониальный илеус с перфорацией кишки и перитонитом может развиться внутриутробно.
В печени (см.) наблюдается диффузная жировая инфильтрация. Повышение вязкости желчи может привести к холестазу с постепенным развитием холестатического гепатита, а также формированием вторичного билиарного цирроза. В происхождении цирроза играют роль также общие обменные нарушения (дефицит аминокислот, липидов, жирорастворимых витаминов); билиарный цирроз может развиться внутриутробно.
Рис. 2. Макропрепарат легких (фронтальный разрез) при муковисцидозе: стрелками указаны множественные абсцессы, окруженные разрастаниями соединительной ткани.
Застой вязкой мокроты в бронхах приводит к обтурационным ателектазам, компенсаторной эмфиземе и вторичному инфицированию. Развивается хрон, бронхит (см.), гнойные бронхоэктазы, хрон, пневмония с абсцедированием (рис. 2).
Клиническая картина
Признаки заболевания могут отмечаться уже в период новорожденности, наиболее выраженная картина болезни в этот период — мекониальный илеус (см. Непроходимость кишечника, у детей), осложнением к-рого является перфорация кишечника и мекониальный перитонит.
По данным М. А. Фадеевой (1977), у 80,3% детей с М. начальные признаки заболевания появляются на первом году жизни. При наличии хорошего, даже повышенного, аппетита дети не прибавляют в весе, могут отставать в росте; тонус мышц и тканей снижен. Отмечается сухость во рту, слюна вязкая; живот вздут, увеличен в объеме, характерен обильный стул, кал резко зловонный, блестящий, светлый, часто серого цвета с большим количеством жира — стеаторея (см.); на пеленках плохо отстирываемые жирные пятна. Могут быть и запоры, но кал и в этих случаях жирный и зловонный. В ряде случаев при наличии запоров у детей, больных М., наблюдается гепатомегалия, выпадение прямой кишки. Нарушаются обменные процессы — отмечаются гипопротеинемия, гиповитаминоз. Нередко заболевание сопровождается нарушением процесса всасывания в кишечнике и пиелонефритом. При рентгенол, исследовании кишечника обнаруживают картину псевдоасцита (расширенные петли кишечника с горизонтальными уровнями содержимого).
Изменения со стороны легких проявляются в виде приступообразного кашля, иногда коклюшеподобного характера. Отмечается постоянная одышка и цианоз.Грудная клетка бочкообразная, ногтевые фаланги имеют форму барабанных палочек. Хрон, гипертензия малого круга кровообращения, обусловленная хрон, воспалительным процессом в легких, ведет к формированию легочного сердца (см.) и сердечной недостаточности (см.) Часто возникают ателектазы (см.) с развитием хрон, пневмонии (см.) и бронхоэктазов (см.). Сгущение секрета в слизистых железах полости носа и придаточных (околоносовых, Т.) пазухах обусловливает развитие хронического ринита, синусита с полипозом. Рентгенологически легочные поражения при М. характеризуются распространенностью и большой пестротой, обусловленной сочетанием перибронхиальных инфильтративных и склеротических изменений на фоне выраженной эмфиземы.
Исходя из указанных симптомов заболевания, времени их проявления и преимущественного поражения тех или иных систем выделяют ряд клин, форм М.: мекониальный илеус новорожденных; легочная форма; кишечная форма; смешанная форма с одновременным поражением жел.-киш. тракта и бронхолегочной системы. Отмечаются также стертые фогмы.
Диагноз
Диагноз основывается на клинических данных, а также результатах Снохим. обследования. Ранняя диагностика М. возможна при массовом обследовании новорожденных путем применения скрининг-тестов. Одним из таких тестов является определение повышенного количества альбумина в меконии как проявления нарушенного всасывания. Диагностическая информативность метода, по данным Брея (Р. Т. Bray), составляет 79%, частота ложноположительных результатов не превышает 5%. Обнаружение повышенного содержания альбумина в меконии служит основанием для дальнейшего углубленного обследования детей. Для постановки диагноза используют также следующие тесты: потовые (определение концентрации ионов натрия и хлора в поте, полученном методом пилокарпин-электрофореза, либо определение концентрации ионов брома в поте после пероральной нагрузки бромистым натрием), определение концентрации ионов натрия в кусочках ногтевых пластинок, тест для определения активности трипсина в кале (растворение или переваривание эмульсионного слоя на рентгеновской пленке фильтратом кала в различном разведении— рентгенопленочный тест) и др.
Наиболее диагностически достоверным и надежным методом выявления М. служит по то вый тест с обнаружением натрия и хлора; патогномоничным для М. считается содержание натрия и хлора в поте св. 40 мэкв/л у детей грудного возраста и св. 60 мэкв/л у детей более старшего возраста. Диагноз М. вероятен при высокой концентрации натрия в ногтевых пластинках.
Дифференциальный диагноз муко-висцидоза представляет известные трудности, особенно на ранних этапах заболевания или при наличии стертых форм болезни. Мекониальный илеус приходится дифференцировать с болезнью Гиршспрунга (см. Мегаколон) и другими пороками развития жел.-киш. тракта. У детей раннего возраста необходимо исключить дисахаридазную недостаточность (см. Мальабсорбции синдром), целиакию (см.), галактоземию (см.). Следует иметь в виду, что клин, картина, сходная с проявлениями М., наблюдается при врожденной гипоплазии поджелудочной железы, а также при дефиците трипсина (см. Поджелудочная железа). Легочную форму М. у детей раннего возраста приходится дифференцировать с коклюшем, бронхиальной астмой (см.).
Лечение
При лечении больных особое внимание должно быть обращено на дието-, ферменто- и и витаминотерапию. Рекомендуется бедная жирами и богатая белками диета. Назначают ферментные препараты, улучшающие функцию поджелудочной железы и жел.-киш. тракта (панкреатин, панзинорм форте, полизим, фестал и др.). В процессе лечения определяют содержание жира в кале, кривую веса тела ребенка.
Для разжижения слизистого секрета и улучшения его оттока назначают ацетилцистеин. При гипопротеинемии проводят переливание плазмы, альбумина. При большом дефиците веса тела применяют препараты анаболических гормонов. Показаны витамины группы В. Лечение мекониального илеуса оперативное.
Прогноз
Прогноз зависит от времени появления первых симптомов М.: чем раньше они возникают у ребенка, тем неблагоприятнее прогноз. По данным ряда исследователей, в 50-х гг. 20 в. 50% детей, страдающих М., умирало в грудном возрасте; с внедрением новых методов лечения продолжительность жизни больных значительно увеличилась.
Профилактика
Каждый плохо развивающийся ребенок, страдающий хрон, пневмонией с частыми обострениями и хрон, расстройствами функции пищеварения, должен быть обследован на М. Дети, больные М., должны находиться под диспансерным наблюдением. Необходимо следить за характером стула и весом ребенка. Периодически (1 раз в 3 мес.) проводят копрологи-ческие исследования для коррекции дозы ферментных препаратов. Два-три раза в год (весной и для профилактики обострения процесса) назначают курсы витаминотерапии.
Библиография: Бадалян Л. О., Таболин В. А. и В e л ь т и щ e в Ю. Е. Наследственные болезни у детей, с. 236 и др., М., 1971; Башляева 3. А. и др. Массовое обследование новорожденных на муковисцидоз, Вопр. охр. мат. и дет., т. 23, № 6, с. 75, 1978; Врожденные и приобретенные энзимопатий, под ред. Т. Ташева, пер. с болг., с. 207, М., 1980; Квачадзе И. М. и др. Кли-нико-морфологическая характеристика му-ковисцидоза у новорожденных, Педиатрия, № 5, с. 31, 1978; Рачинский С. В., Таточенко В. К. и Капранов Н. И. Муковисцидоз у детей, М., 1974, библиогр.; Фадеева М. А. и др. Морфологические изменения при муковисцидозе у детей, Вопр. охр. мат. и дет., т. 19, № 7, с. 44, 1974; М ихов X. и Антова В. Муковисци-доза, Пловдив, 1977; Andersen D. H. Cystic fibrosis of the pancreas and its relation to celiac disease, Amer. J. Dis. Child., v. 56, p. 344, 1938; Bray P. T. a. o. Sweat testing for cystic fibrosis, Arch. Dis. Child, v. 53, p. 483, 1978; F a r b e r S. The relation of pancreatic achylia to meconium ileus, J. Pediat., v. 24, p. 387, 1944; Meyer H. TheraDie der pulmonalen Symptome der cystischen Fibrose, Mschr. Kinderheilk., Bd 126, p. 168, 1978; Pas-s a r g e E. Cystische fibrose, ibid., p. 172.
В. А. Таболин; Т. E. Ивановская (пат. ан.).
Источник